一、医疗器械产品在美国上市许可的三种申请类型
- 直接进行公司和产品注册/登记,无需向FDA递交产品安全性和有效性报告,适用于大部分I类和少部分II类产品;
- 先获得FDA 510(K)/PMN批准,然后进行公司和产品注册/登记,适用于大部分II类和少部分I、III类产品;
- 先获得PMA批准,然后进行公司和产品注册/登记,适用于大部分III类和少部分II类产品。
二、什么是510(K)?
510(K)来源于美国《联邦食品、药品和化妆品法案》第510条(K)条款,具体指医疗器械产品上市前的预通告制度(PMN: Pre-Marketing Notification)。对适用的医疗器械产品,在美国上市前,申请人需要向FDA递交的一份安全性和有效性的论证报告,论证该器械产品和美国同类产品(等价器械,Predicate Device)在安全性和有效性方面的相似程度,批准时会获得一个510(K)号码。也就是说申请人必须比较一个或更多的类似合法上市的器械,证明申请上市的器械与已经在市场上销售的器械同样安全有效,即具有『实质性等同(substantial equivalence, SE)』。
在获得FDA的许可通知之后,申请人才可以进行产品注册和合法上市。FDA会详加审查此产品是否与申请者所指定的产品即等价器械具有实质性等同。
三、510(K)的审核流程
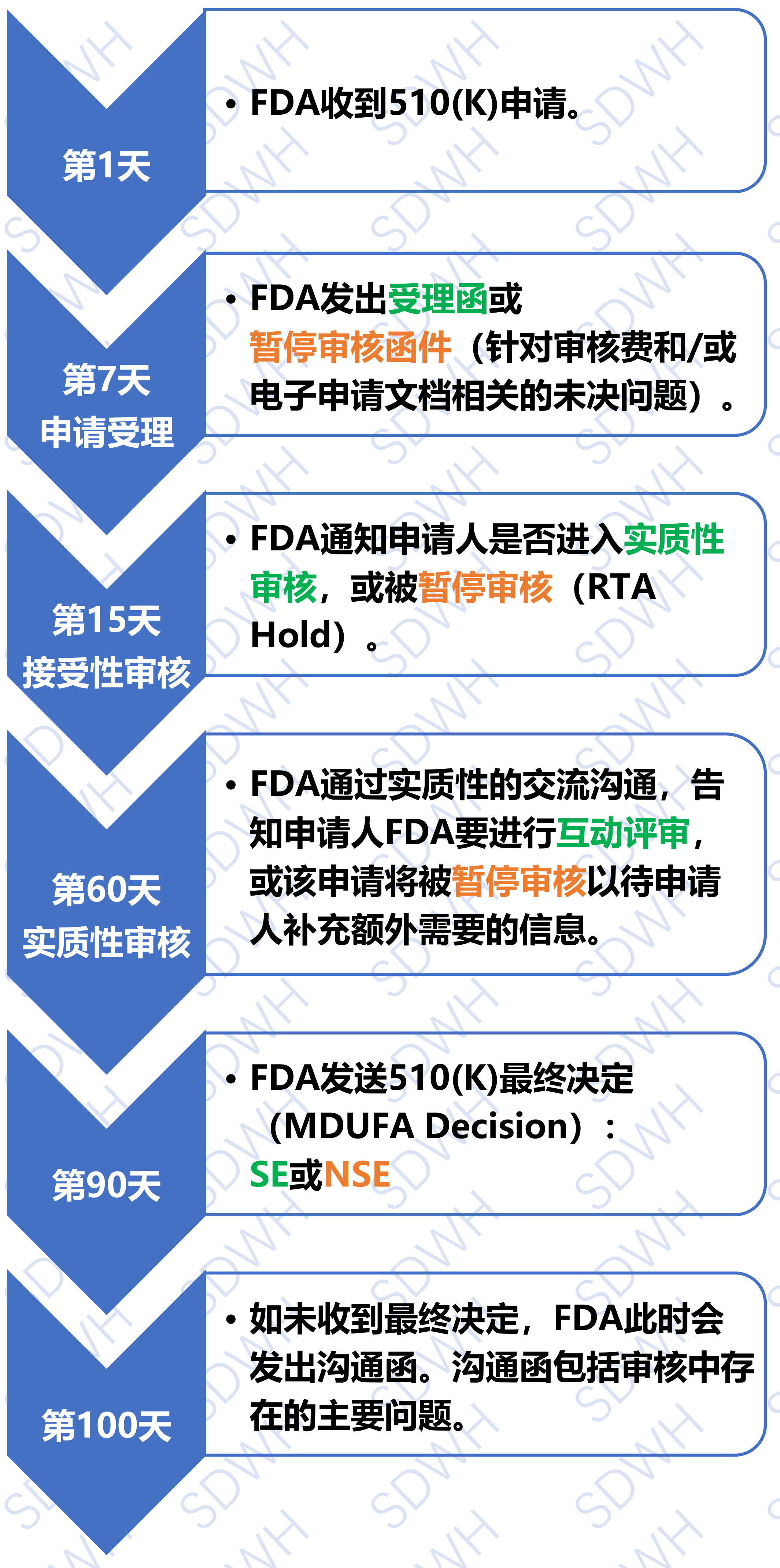
注:
- 流程上的时间是自然日;
- 需要补充信息时,审核会暂停,申请人需在180天(自然日)内做出回复;
补充的信息资料需同时递交纸质版和电子版。
在申请人收到符合SE命令之前,该器械不能在美国市场上销售。FDA通常是在90天内根据申请人提交的信息判断是否符合SE。如果FDA判定该器械为NSE,申请人可以:
- 用新数据重新提交一份510(K)文件;
- 通过De Novo分类流程申请I类或II类重新分类申请;
- 提交上市前批准申请(PMA)。
四、实质性等同(substantially equivalent, SE)判定标准
- 相同用途、相同技术特征(指材料、设计、能源或其他特征);
- 相同用途、不同技术特征,但具有相同的安全性(safety)及功效性(effectiveness),而且不会产生与等价器械不同的有关安全性和有效性方面的问题。
FDA 510(K)的论证主要包含两部分:产品本身的相关论证和信息;与等价器械的安全性/有效性比较论证。
五、510(K)的申报资料
申报资料一般包括以下部分:
- 申请函、目录(文件清单,含附件)、真实性保证声明(FDA有标准范本)、注册号码(如未注册,须注明)、510(K)摘要或声明;
- 产品信息:器材名称、分类、性能标准、产品标识;
- 实质等同性比较(SE);
- 产品描述:产品的预期用途、工作原理、动力来源、零组件、照片、工艺图、装配图、结构示意图等;
- 产品的安全性与有效性,包括各种设计、测试资料;
- 生物相容性;
- 色素添加剂(如适用);
- 软件验证(如适用);
- 灭菌(如适用),包括灭菌方法的描述、灭菌验证产品包装和标识等。
注:不适用的项目可以选择不提供。如,无源产品无需提供软件技术文件和软件测试结果,多数成熟产品无需提供临床试验结果。
六、510(K)常见问题
 哪些情形需要递交510(K)?
哪些情形需要递交510(K)?
- 非豁免的、首次在美国上市的医疗器械产品,;
- 对于已上市器械提出不同的使用目的;
- 已上市器械发生重大变更,此变更能够严重影响器械的安全性或有效性的。
更多信息戳此链接??Is a new 510(k) required for a modification to the device?
关于经销商。以自己公司的名称分销制造商的产品,需要提交510(K)吗?
关于国外厂商。
- 国外制造商可以直接向FDA提交510(K)。方便起见,国外制造商应指定美国境内的自然人或在美国有办公处所的实体法人作为代理人,由其负责紧急情况和日常事务交流。
申请人需要在提交510(K)之前进行器械注册登记吗?
- 不需要。如果是一家新公司并且不生产任何医疗器械产品,则应在制造和销售器械后30天内进行注册。提交510(K)时应说明当前未注册登记厂商信息。
关于质量体系。在提交510(K)之前,是否需要对申请上市器械进行质量体系核查?是否需要在510(K)申请中提供申请上市器械符合质量体系文件的要求?
- 不需要。FDA 510(K)是文件审核;只针对产品,不涉及质量体系。但申请人应随时准备接受FDA检查。
关于第三方510(K)审核
- 第三方510(K)审核程序是FDA为医疗器械注册申请人提供的一个自愿的替代性的审核程序。该程序允许有资质认可的第三方510(K)审核机构(Third Party (3P510k) Review Organization)审核规定的中、低风险的医疗器械,以提高510(K)的审核效率。
详情戳此链接??您家的器械可以申请FDA第三方510(K)审核吗?
参考资料:
- 【苏大检测】公开课第1讲之美国FDA医疗器械法规和实施
- 美国医疗器械上市前监管概述与启示.中国医疗设备.2020,35(03):160-170.
- 如何进入美国医疗器械市场——医疗器械企业注册、产品列名与510(K)申请.中国医疗器械信息.2003,(4):33-35.
- https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm
- https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm
- https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/premarket-notification-510k