之前小编已为大家带来《【医械检测科普系列之17】降解产物与可沥滤物毒代动力学研究设计》,本期推出的是16886中第17部分可沥滤物允许限量的建立相关内容,包括特定可沥滤物可耐受摄入量(TI)的建立、可耐受接触(TE)的计算、可行性评价、受益评价及报告要求等。想要复习过往内容的小伙伴请戳这里《【医械检测科普系列之17】降解产物与可沥滤物毒代动力学研究设计》。
【医械检测科普系列】专题是小编根据16886系列标准精心打磨,用心锤炼而来,旨在帮助大家更全面准确地理解标准的内容。请大家多多支持哦!小伙伴们,还在等什么?请为小编点赞+收藏+关注+在看!收下小编最诚挚的祝福!
医疗器械在临床使用中,有害的可沥滤物将释放进入人体。这时应根据特定可沥滤物的健康风险分析并在对损害健康具有适当保护程度的水平上建立允许接触限量。
GB/T 16886 的本部分提供了运用健康风险数据计算最大允许限量的方法,其目的是获得标准的运用及未建立标准的限量的适当评估。本部分描述了一个系统过程,通过该过程,医疗器械中毒害物质产生的确定风险被量化。
GB/T 16886 的本部分规定了医疗器械可沥滤物允许限量的确定方法,小编已为您梳理出其中的重点内容:
一、允许限量建立的一般原则
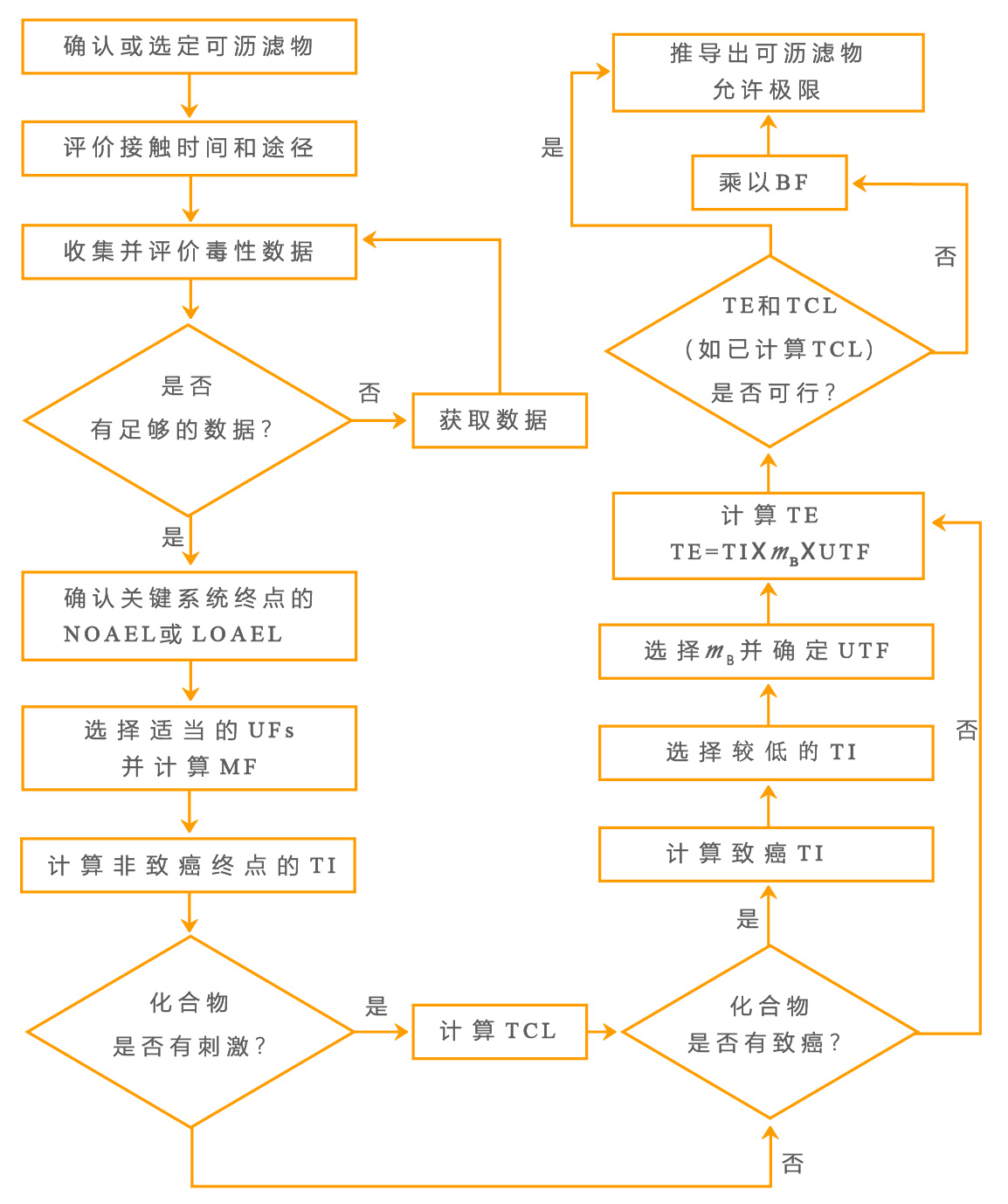
►医疗器械中确认的可沥滤物允许限量建立的过程(见图1),其构成为:
1、利用下列方式评价与可沥滤物相关的生物学风险
——收集数据并确认临界健康终点;
——根据特定接触时间和接触途径确定可耐受摄入量(TI);
——若刺激作用是一个合适的终点,确定可耐受接触水平(TCL);
2、利用下列方式确定病人对可沥滤物的可耐受接触(TE)
——确定病人合适的体质量(ms);
——根据医疗器械的应用因子(UTF)修正可耐受接触摄入量和体重的乘积;
3、适宜时确定可行性和应用受益。如果可行性评价确定该TE在技术和经济上可行,则该TE就成为允许限量。如果TE在技术和经济上不可行,则按照要求建立允许限量的过程进行受益评价,从而进一步修正该TE。
图1、可沥滤物允许限量的建立
►可沥滤物允许限量的建立应由富有理论知识和实践经验以及具有科学有效数据做出正确判断能力的人员通过相应专业判断予以实施。
►医疗器械的安全性要求无不可接受的健康风险。健康风险可接受性的评估需要研究和平衡几个复杂的因素。风险性评估的可信性与被评价数据的数量和质量有关。
►许多情况下,毒理学数据有充分的一致性,对于可沥滤物的一个使用类别或者最具代表性的毒理学作用的接触途径来说,允许使用最低TI。
►建立允许限量的第一步是有害健康物质的确认。一旦有害物质被选定,允许限量的建立过程应以可耐受摄入量建立为始。
二、特定可沥滤物可耐受摄入量(TI)的建立
总则
毒理学数据的评审为建立无可观察到的不良反应水平(NOAEL)提供了必要信息。然后将修正因子的方法应用到无致癌终点的数据中以便能建立适宜的可耐受摄入值。修正因子或定量的方法均可用于确定源自致癌数据的可耐受摄入量。
TI计算的接触因素
►所用数据
1、病人接触可沥滤物的持续时间;
2、病人接触可沥滤物的常规途径。
►接触持续时间因素
1、对特定器械的接触持续时间运用GB/T 16886.1规定分类并进行相应的数据分析。
2、若是对所有医疗器械建立可沥滤物TI, 应在该可沥滤物生物学作用基础上,计算持久接触TI,需要时,以长期接触和短期接触的TI作为附加约束。若是对特定持续时间类别的一个或一类器械存在的可沥滤物建立TI时,应在可沥滤物生物学基础上,建立该持续时间类别的TI,必要时,以较短接触时间类别的TI作为附件约束。
3、对特定的类别,如果没有有效的数据建立TI,应使用较短期的研究数据和较大的修正因子。
4、若器械有多种使用类别,则应按最严的分类建立TI。
►接触途径因素
当对所有接触途径建立器械可沥滤物TI时,尽可能按照GB/T 16886.1所给定的接触时间类别对每一个可能的接触途径计算TI。当对于特殊途径没有有效数据时,可用其他有数据途径的TI作为该无数据途径的TI。
数据的收集和评价
►一旦选定要评价的可沥滤物,应收集相关的有效数据,这些数据包括:
1、化学和物理性质;
2、产生和使用;
3、药理作用;
4、毒代动力学(吸收、分布、代谢和排除);
5、毒理学;
6、对人类的作用。
►用于设置限量的数据应该是高质量并高度相关的。长期数据可用于建立短期限量。人类数据则优于动物数据。
►确认重要不良作用以及建立这些作用的NOAELs的数据需要进行评价。适宜时,应对来自多种接触途径的数据给予评价。在仅有一个接触途径情况下,虽然可以考虑其他途径的数据,但与该途径相关的数据是最合适的。
►在考虑人类预期接触途径时,应确认作为设置限量基础的最直接相关的不良作用以及产生这些反应的剂量。剂量选择的基本原则应形成文件。
设置非致癌终点的TI
►总则
1、对每一个预期接触途径和期限,TI应根据已确定的NOAEL,LOAEL或其他数值计算。每一个TI计算都应考虑确认的损害的严重程度和风险分析中所固有的不确定性。
2、无论何时,应尽可能使用修正因子的方法计算各TI。该方法是使用专业判断基础上确定的不确定因子和提供一个相对于最相关不良作用的可接受的安全程度。
3、限量应根据预期使用人群最宽分布来建立,应以与预期使用接触相关的人为评价依据。
4、致癌可沥滤物TI确定的步骤
5、可沥滤物的致癌性一旦确定,致癌TI应与非致癌终点的TI值一起评价,以便确定TE计算中使用合适的持久接触TI。
6、建立各TI时,从健康危险的严重程度观点出发,应考虑适宜的安全保证程度。
致癌终点的TI的设置
►致癌可沥滤物TI确定的步骤
1、可沥滤物的致癌性一旦确定,致癌TI应与非致癌终点的TI值一起评价,以便确定TE计算中使用合适的持久接触TI。
2、对致癌的可沥滤物,证据加权测试的适当方法用于致癌TI的确定。证据加权测试包括回答下列问题:
——该材料是否是遗传毒致癌物?
——肿瘤类型是否是与人类相关?
——是否有生物倾向数据支持对人类的推断?
——流行病学信息是否支持与人类相关?
►通过证据加权测试的内容选项
如果证据加权测试表明该材料是遗传毒致癌物以及癌症生物鉴别观察的肿瘤类型与人类相关,并且生物倾向和/或流行病学支持与人类的相关性。应使用下列方法之一:
1、使用10-4显著风险水平的统计模式,根据量化风险评定程序来确定癌症TI;
2、不确定癌症的TI,尽可能合理地减少病人的实际接触并利用风险管理程序加强 癌症风险的管理。
►证据加权测试失败或不可靠时的程序
1、如果证据加权测试失败,则应使用修正因子的方法。如果证据加权测试不可靠,应使用修正因子和量化风险评估的方法来确定癌症TI。
2、无论何时,应尽可能使用生物药理动力学模型(PBPK)评价源自该靶器官的剂量而不是应用剂量,风险计算也使用该剂量而不是应用剂量。
可耐受接触水平(TCL)的建立
►总则
刺激数据的评审为决定是否需要考虑刺激,和必要时建立无刺激作用水平(NIL)提供了必要的信息资料。一旦决定要获取一个NIL,就要使用修正因子的方法以便获得可耐受接触水平。
►TCL计算的接触考虑
1、任何通过与人体组织直接接触能产生刺激作用以及从指定器械使用样品表面缺口产生的可沥滤物可能都需要可耐受接触水平(TCL)。
2、对于多次组织接触应用可能需要可耐受接触水平(TCL)。
►刺激终点的TCL设置
1、对每一个相关接触组织,都应根据无刺激作用水平(NIL),最小刺激作用水平(MIL)或其他相似的水平计算一个TCL。每一个TCL计算时应考虑从多种接触到没有刺激作用的浓度的刺激程度,只要有这些数据存在。应使用更改因子的方法计算TCL。这种方法要与根据专业判断所确定的不确定因子的使用相结合,以便给出对刺激的可接受的安全裕度。
2、应根据特定用户人群的最广泛分布来建立刺激限量。如果预期不是一般使用,就要使用器械预期应用的人群。
3、不确定因子的选择应包含几种考虑:
——不确定因子4(UF4)
UF4说明人体间个体差异,推导TCL时应考虑UF4。最好有评价人体差异的真实数据。
——不确定因5(UF5)
UF5表示非人物种获取的数据向人类的推断,UF5应考虑非人物种与人类的固有差异。
——不确定因子6(UF6)
UF6表示实验数据的质量和相关性,用MIL代替NIL时,可能需要3或更大的UF6。
混合物的危险评定
1、大多数病人都是同时在接触医疗器械释放的多种化合物,这种同时接触可能增加或降低任一给定被研究物质的毒性。然而医疗器械释放化合物的速度低于这些化合物各自的TI值时,混合成分中发生的增强作用的可能性不大。
2、如果一个器械沥滤物的化合物以常见毒理学作用机制起作用或者其结构类似于其他的一个物质(如肽酯、丙烯酸盐、甲基丙烯酸盐)并且病人接受的这些化合物的剂量低于每一个化合物各自的TI值时,可假定以添加的方式产生作用即两种或多种介质综合作用等于各介质单独作用的总和。因此用危害指数(HI)方法来评价接触混合物产生不良作用的可能性。
可耐受接触(TE)的计算
►总则
1、已知一种可沥滤物的TI,有必要调整合适的TI以确定接触量将是可耐受的,这就要考虑器械的使用方式和接触来源于其他医疗器械的该可沥滤物的可能性。
2、应评价下列因素,以确定适宜的体质量和用来确定可耐受接触(TE)的应用因子(UTF):
1)接触器械的特定群体;
2)接触群体的主要体质量;
3)器械预期使用模式;
4)病人从多种医疗器械接触同一可沥滤物的可能性。
接触群体
►体质量
TE应根据使用器械的群体选择体质量,尤其预期用于独特敏感群体(如婴儿)的器械,要有特别的考虑。
►婴儿和儿童的专用器械
对婴儿使用的器械,应考虑材料排出的主要途径的可能不成熟和在体内的较大蓄积。计算预期用于婴儿的医疗器械的各TI时,源自婴儿接触有害材料的数据更为可取。当得不到这样的数据来计算Tls时,以成人数据计算的TIs被用于计算TE。
从使用模式计算应用因子
►总则
1、可耐受摄入量(TI)和体质量之积通过乘以应用因子(UTF)来予以调整。
2、医疗器械的一般使用模式(包括其用作治疗体系的一部分)应由其应用群体决定。应用因子的推算应尽可能考虑医疗器械的预期使用模式。
►比例接触因子
医疗器械不能在一个接触类别的全部接触时间内使用,可以用调高一个应用因子(UTF)来说明这种情况。为了方便,从器械预期产生的实际接触时间所占接触类别的比例来计算PEF。
可耐受接触(TE)
考虑器械使用方式以调整可耐受摄入量(TI)。可耐受接触TE是可耐受摄入量、体质量和应用因子的乘积。
四、可行性评价
►可行性表示制造商或再加工商达到可耐受接触的能力。包括两部分:
1)技术可行性;
2)经济可行性。
♦技术可行性表示不考虑成本获得一个器械或一个类型的器械的可耐受接触。
♦经济可行性表示在不考虑器械不合理经济因素的前提下去漫展可耐受接触的能力。在选择允许限量时,应考虑影响人体健康的预防、增进或改善的成本和效用。
►如果获得可耐受接触(TE)是可行的,受益评价不需再进行,受益因子设为1且允许限量与可耐受接触相同。如果可耐受在技术或经济上是不可行的,应进行受益评价。受益的考虑说明应形成文件。
五、受益评价
►在应用医疗器械风险性评估中,可作出这样的期望,即所有医疗过程都伴有健康风险,而医疗使用的风险要与使用它所带来的健康受益进行衡量。
►假如可沥滤物是从材料或加工过程产生的毒性化合物,而又没有其他代替材料或加工方来加以避免,应考虑器械使用所带来的健康受益。在允许限量计算中使用的受益因子的必要性和大小的理由应形成文件,这种情况下,允许限量就是TE和受益因子(BF)的乘积。
六、允许限量
►在计算各TE并根据可行性和受益对它们进行修正后,对各TE计算允许限量。要求满足所有允许限量。
►允许限量也可用毫克/每器械表达。
七、报告要求
总则
►建立医疗器械可沥滤物允许限量所用的任何资料和说明合理的文件都要充分。尽可能地提供如下形式的报告,每一个器械的报告或者每一种物质的报告或者根据个体情况的推断性报告。报告内容尽可能简短。
►报告应包含下列信息:
1)可沥滤物的确定;
2)问题器械的简短描述;
3)关键的NOAEL, LOAEL, NIL和(或)可沥滤物其他终点,每一项都应提到;修正因子的选择和论证(UF1、UF2、UF3等的论证);
4)非致癌性TI;
5)如适当,致癌性TI;
6)如适当,TCL;
7)UTF和它的理由(即CEF和PEF的理由);
8)TF和它的理由;
9)涉及使用的所有相关数据的可行性评价的综述;
10)如适宜,BF的选择及其论证,附所有的关键数据理由;
11)可沥滤物的允许限量;
12)根据GB/T 16886本部分叙述的方法计算的允许限量作用的描述。
原文来源:GB/T 16886.17-2005
全文整理:苏州大学卫生与环境技术研究所
声明:如果您认为我们的内容或来源标注与原文不符,请告知我们,我们将与您积极协商解决。谢谢大家的关注!